





Introduction
Sickle cell anemia is a severe inherited blood disorder that affects the structure and function of red blood cells. Unlike normal disc-shaped cells, those in sickle cell anemia become stiff and “sickle-shaped,” leading to a cascade of complications, including poor oxygen delivery, pain, increased risk of infection, and long-term organ damage.
This article explains the condition in clear sections — from what it is, to how it affects the body, what symptoms it causes, how it is diagnosed and managed, and what treatments are available.
What Is Sickle Cell Anemia?
Sickle cell anemia is a genetic blood disorder characterised by the production of abnormal haemoglobin called haemoglobin S. Haemoglobin is the protein in red blood cells that carries oxygen throughout the body.
How the Cells Change:
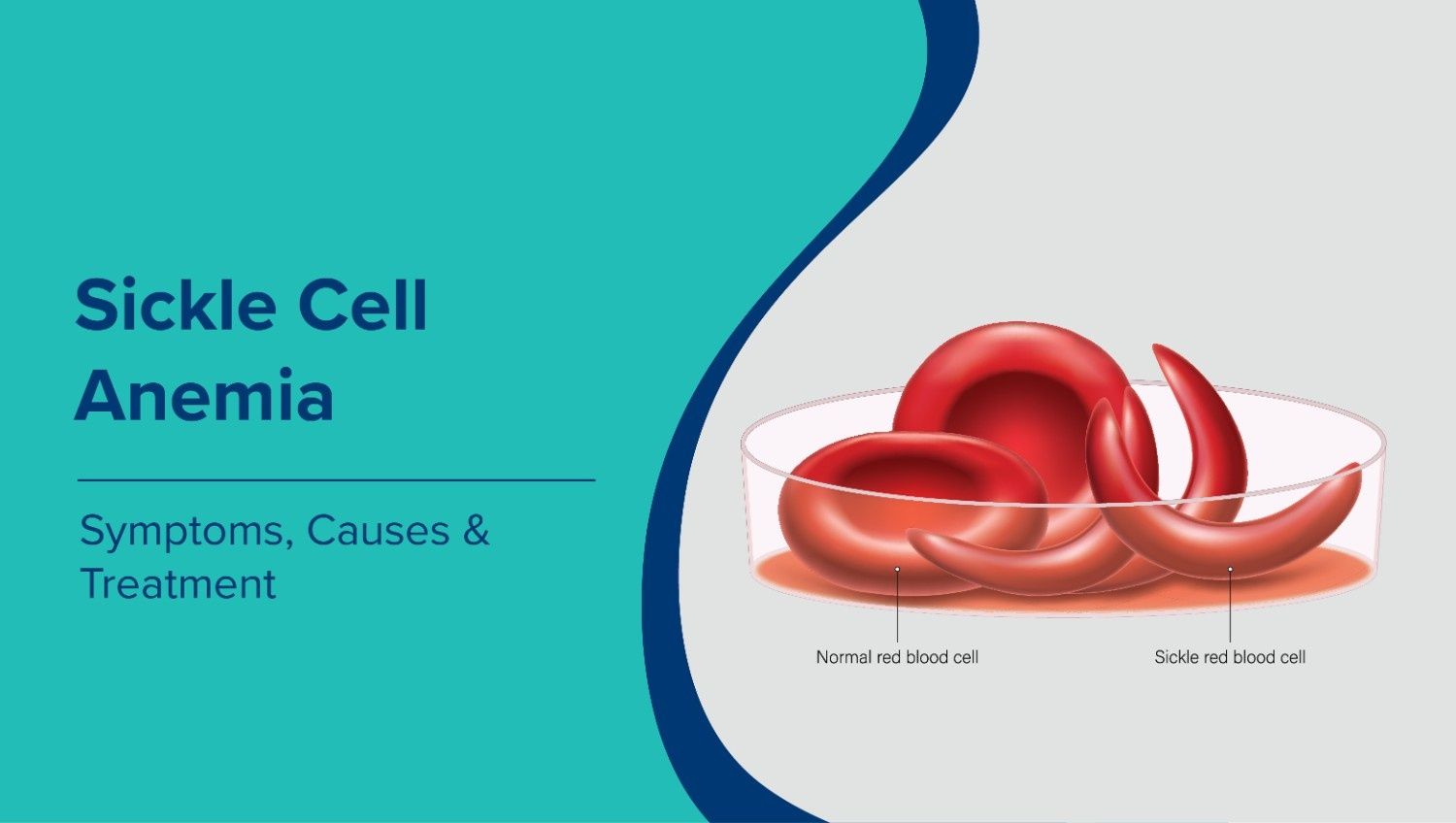
- In a healthy person, red blood cells are round and flexible, allowing them to travel easily through small blood vessels.
- In sickle cell anemia, the haemoglobin mutation causes red blood cells to become rigid and crescent-shaped (“sickled”).
- These sickle cells are less able to carry oxygen and are prone to getting stuck in narrow arteries, leading to blockages, pain, and organ damage.
Who Gets It:
It is a genetic disorder passed down from parents. A child needs to inherit the sickle cell gene from both parents to have the disease. If only one gene is inherited, the child might carry the trait but will not have the full disease.
How the Disease Develops—Pathophysiology Explained
Understanding how sickle cell anemia affects the body at a cellular level helps explain its wide range of symptoms and complications:
Genetic Mutation
The disorder stems from a mutation at a specific point in the hemoglobin β-globin gene. This mutation causes changes in the hemoglobin molecule, making it likely to aggregate when oxygen levels are low. This aggregation alters the shape of red blood cells.
Sickling of Red Blood Cells
When oxygen levels drop, such as at high altitude or during dehydration, hemoglobin S polymerizes, causing red blood cells to become rigid and sickle-shaped. These abnormal cells are short-lived and break down prematurely, resulting in chronic anemia.
Vascular Blockage and Tissue Damage
Sickle cells can obstruct small blood vessels (vaso-occlusion), leading to decreased blood flow to tissues. This blockage reduces oxygen supply, resulting in painful episodes and contributing to long-term organ damage.
Hemolysis and Anemia
Since sickled cells break down more quickly than normal red blood cells, the body struggles to replace them fast enough, resulting in anemia a deficiency of healthy oxygen-carrying cells.
What Are the Signs and Symptoms Associated with Sickle Cell Anemia
The symptoms are varied and can range from mild to severe. People with the disease may have periods without symptoms, interrupted by painful episodes or complications.
Key Symptoms Include:
Chronic anemia
The shortened lifespan of sickled cells causes symptoms such as fatigue, paleness, and weakness because tissues lack sufficient oxygen.
Pain Crises (Vaso-occlusive Episodes)
Severe pain episodes happen when sickle cells obstruct blood flow in small vessels. This pain can impact the back, chest, arms, legs, and abdomen. Such crises may last from hours to days and sometimes necessitate hospitalization.
Dactylitis
Swelling and pain in the hands and feet, especially in infants and young children, are caused by blocked blood flow in the extremities.
Jaundice
Yellowish discoloration of the skin and eyes due to the rapid breakdown of red blood cells.
Increased Infections
The spleen often becomes damaged or non-functional, impairing the body’s ability to fight infections. Individuals with sickle cell anemia are more susceptible to serious infections.
Stroke and Neurological Problems
Blocked blood flow to the brain can lead to stroke in children and adults alike. This is one of the most serious complications.
Acute Chest Syndrome
A life-threatening lung problem that presents with chest pain, fever, and difficulty breathing. It is one of the leading causes of hospitalisation and death in people with sickle cell disease.
Complications And Long-Term Effects
Sickle cell anemia is a systemic disease, meaning it can affect many body organs:
Organ Damage
- Kidneys (leading to chronic kidney disease)
- Heart and lungs (pulmonary hypertension)
- Bones and joints (avascular necrosis)
- Eyes (retinopathy and vision loss)
Chronic Pain
Repeated damage to bones and tissues can lead to chronic pain beyond acute crises.
Growth Delay and Developmental Issues
Children with sickle cell anemia may have delayed growth and puberty due to chronic anemia and nutritional deficiencies.
Life Expectancy
Without treatment, severe sickle cell disease may lead to early mortality. With modern care, life expectancy has significantly improved, though it varies widely by region and access to healthcare.
How Sickle Cell anemia Is Diagnosed
Newborn Screening
Sickle cell disease is commonly detected in newborns through routine screening tests, enabling early diagnosis and prompt treatment.
Blood Tests
- Complete Blood Count (CBC): measures overall blood parameters including hemoglobin count, shows anemia.
- Haemoglobin Electrophoresis: identifies abnormal haemoglobin S.
- Genetic Testing: confirms the specific gene mutations.
Early diagnosis helps doctors plan preventive care and reduce complications.
Treatment For Sickle Cell Anemia
Although no single cure exists for all patients, numerous treatments can manage symptoms, prevent complications, and enhance quality of life.
Supportive Care
- Hydration and Oxygen: Staying well hydrated and oxygenated can reduce sickling.
- Pain Management: Pain relief with medications and supportive therapies during pain crises.
Medications
- Hydroxyurea: Widely used to reduce the frequency of pain episodes and acute complications.
- L-glutamine and other agents: Help reduce oxidative stress and prevent sickling.
- Vaccines and Antibiotics: Help prevent serious infections.
Blood Transfusions
- Acute Transfusion: Used during severe crises to quickly increase oxygen capacity.
- Chronic Transfusion: May be given long-term to prevent recurrent strokes and complications.
Bone Marrow or Stem Cell Transplant
It can be curative, especially in children who have a matched donor. Not all patients are candidates due to risks and donor availability.
Emerging Therapies
Recent gene therapy approaches aim to correct the genetic defect itself, offering the possibility of a lifelong cure for some patients. (Cleveland Clinic)
Living With Sickle Cell Anemia
Regular Medical Follow-Up
Consistent checkups help monitor organ function and prevent complications.
Infection Prevention
Stay up-to-date with vaccinations and seek early treatment for infections.
Healthy Lifestyle
Adequate hydration, a nutritious diet, and avoiding extreme temperatures can reduce crisis triggers.
Emotional and Social Support
Chronic illness can affect mental health and quality of life. Support groups, counselling, and family education are essential.
Conclusion
Sickle cell anemia is a hereditary condition that greatly impacts health and daily life. However, advances in medicine, early detection, and new therapies have transformed it from a fatal childhood disease into a manageable chronic condition for many individuals.
Understanding the disease, including its causes, symptoms, complications, and treatments, empowers patients, families, and carers to work with healthcare providers and achieve better health outcomes.





